
DATE2022.01.07 #Press Releases
Deep distributed computing for giant phylogenetic tree estimation
Disclaimer: machine translated by DeepL which may contain errors.
Naoki Konno (Second-year student, Master's Program, Department of Biological Sciences)
Wataru Iwasaki (Professor, Department of Integrated Biosciences, Graduate School of Frontier Sciences / Professor, Department of Biological Sciences)
Nozomu TANIUCHIE (Associate Professor and Canada Research Chair, University of British Columbia / Visiting Associate Professor, Research Center for Advanced Science and Technology, The University of Tokyo)
Key points of the presentation
- We have developed a "deep distributed computing" FRACTAL method (Note 1) to estimate huge phylogenetic trees at high speed from a large number of DNA sequences.
- This method dramatically expands the number of DNA sequences that can be handled by various phylogenetic tree estimation software, and it was shown that this method can be used to estimate huge phylogenetic trees consisting of at least 200 million sequences.
- It is expected to become a key method in a wide range of biological fields, including the elucidation of the evolutionary process of microorganisms and viruses, and the developmental process of animals.
Summary of Presentation
Organisms evolve by changing their genomic DNA sequences, which consist of the four letters A, T, G, and C. Evolutionary biology focuses on the evolution of organisms on the earth. In evolutionary biology, techniques have been developed to infer the evolutionary phylogenetic tree, the genealogy of the evolution of organisms on earth, from their DNA sequences. Recently, this concept has been brought into developmental biology. In particular, projects are underway worldwide to reconstruct the developmental process of the whole body from the DNA mutation information of individual cells obtained from fertilized eggs that have been modified to rapidly accumulate mutations in partial artificial DNA sequences on their chromosomes. On the other hand, existing methods for genealogical inference are computationally time-consuming and memory-intensive, and the number of sequences that can be targeted is limited to about 1 million.
A joint research team led by Naoki Konno, Master's student, and Wataru Iwasaki, Professor, Department of Biological Sciences, Graduate School of Science, The University of Tokyo, and Nozomu Taniuchi, Associate Professor, University of British Columbia, has invented FRACTAL, a "deep distributed computing" method, which can accurately estimate genealogies consisting of over 200 million sequences and can be used for evolutionary phylogenetic tree They have shown that FRACTAL can be used to accurately estimate genealogies consisting of more than 200 million sequences, and to greatly expand evolutionary phylogenetic tree and cellular lineage tracing.
This technology is expected to expand the horizon of biology in a wide range of fields, including large-scale evolution of microorganisms and viruses, and high-resolution developmental processes of animals.
Contents of Presentation
Organisms have evolved by gradually changing their DNA sequences, which consist of the four letters A, T, G, and C. In the process, the DNA sequences of different species have changed little by little. In this process, one species repeatedly diverges into two different species, and the genealogy of past speciation is represented as a "tree" of repeated bifurcations, or an evolutionary phylogenetic tree. Since we can assume that two species with more similar DNA sequences diverged more recently, we can infer their evolutionary phylogenetic trees by comparing the DNA sequences of many species currently living on the earth.
Inspired by this evolutionary phylogenetic tree, recent years have seen the development of technologies for tracing cell lineage, the process by which cells divide during animal development. In such an idea, using the genome editing technology (Note 2), a contrivance is prepared in which mutations are introduced into a short artificial DNA sequence introduced into a chromosome as the cell divides. By obtaining individual animals from fertilized eggs carrying these DNA sequences and analyzing the DNA mutation information of each descendant cell, it is possible to obtain genealogical information on how cell division progressed from the fertilized egg to form the individual animal, as in the case of evolutionary phylogenetic tree estimation.
However, in general, methods to estimate phylogenetic trees from DNA sequences are computationally time-consuming and memory-intensive, and the number of sequences that can be used to estimate genealogies has been limited to about 1 million at the maximum. Considering that the number of eukaryotic species alone is estimated to be 8.7 million, and that microorganisms and viruses are being identified one after another through genome and metagenome analysis, the number of DNA sequences that evolutionary biology will need to handle in the future is expected to greatly exceed this computational limit. In addition, cell lineage tracing during animal development is also expected to face a computational limit in the future, considering that vertebrates are composed of tens of billions to trillions of cells.
In this study, a research group led by Naoki Konno, Master's student, and Wataru Iwasaki, Professor, Department of Biological Sciences, Graduate School of Science, The University of Tokyo, in collaboration with Nozomu Taniuchi, Associate Professor, Department of Biomedical Engineering, University of British Columbia, and their research team, have developed a new method to extend the number of sequences that can be handled by various genealogy estimation software. They have developed FRACTAL, a "deep distributed computing" method that expands the number of sequences handled by various genealogy estimation software. The team hypothesized that monitoring the computational status of all sequences handled by ordinary genealogy software in a sequential manner would be overcomputing for solving many practical problems, and demonstrated this in the form of an implementation of FRACTAL.
FRACTAL can incorporate any genealogy estimation software as a plug-in; FRACTAL first randomly samples only a subset of the large number of input DNA sequences and reconstructs a small genealogy tree of them (small computational effort) (Figure 1).
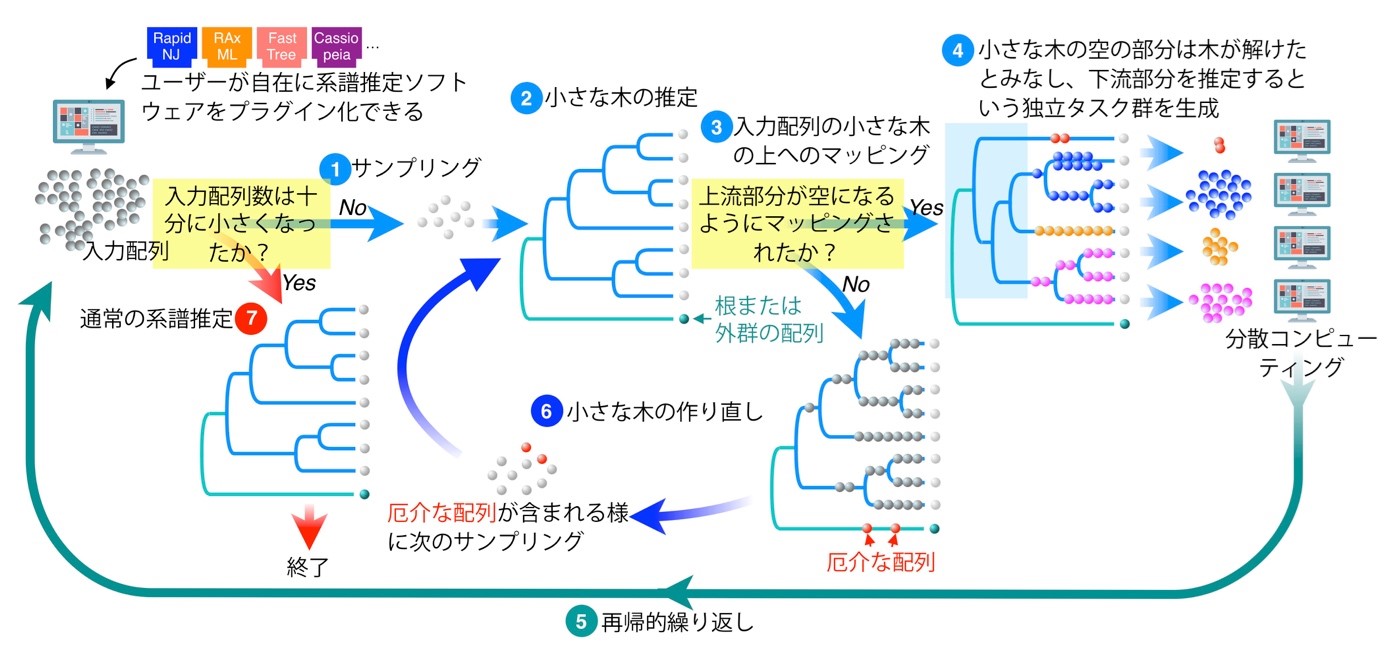
Figure 1: Overview of the process of estimating a giant phylogenetic tree with FRACTAL.
Next, each of all input DNA sequences is estimated as to which position on that small tree it is closest to (small computational effort). As a result, if an upstream portion of the tree appears where no input sequence maps in that small tree, we consider this upstream portion to have been "solved". Since the remaining input sequences are mapped by hanging from the branches of the tree in the upstream portion, it is sufficient to estimate the downstream tree independently for each group of sequences hanging from each branch. Thus, the same process can be repeated with different independent computers for this split group of sequences (if no such "empty" upstream portion appears, then sampling with the bias learned from this result is repeated until a small tree with an "empty" upstream portion is obtained). In this way, FRACTAL enables the estimation of huge genealogies by adopting a form in which a computer performs a small task and then allocates the remaining tasks to other computers, and the computers to which the task was allocated also allocate tasks to another large number of computers.
In this study, we applied this method to a variety of DNA sequence data and showed that FRACTAL is capable of various types of giant genealogy estimation. For example, we generated DNA sequences with over 235 million mutations introduced by simulation and ran FRACTAL using a 300-node computer. The results showed that FRACTAL was able to reproduce the sequence generation process of the simulation with an accuracy of more than 99.8% within 32 hours (Figure 2).
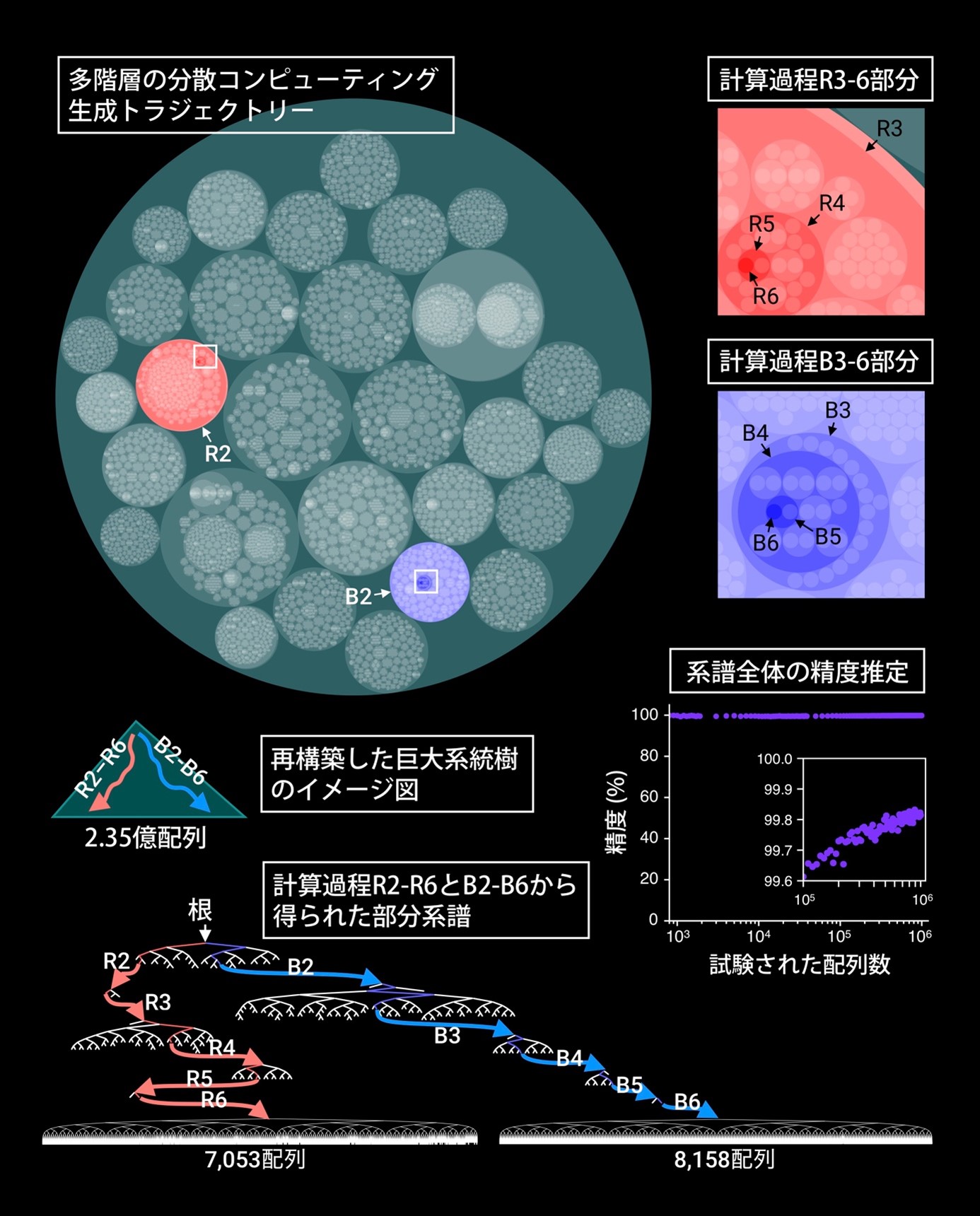
Figure 2: Phylogenetic tree reconstruction of 235 million sequences using FRACTAL. In a multi-tiered distributed computing generation trajectory, one circle represents a single computational job, and the distributed computing tasks it generated are indicated by the next tier of circles. Since the accuracy of the entire huge genealogy cannot be directly measured, the accuracy was estimated from the accuracy values of partial genealogies.
In addition, to verify the applicability of the method to DNA sequences of natural organisms, we also verified that the pseudo-giant evolutionary phylogenetic tree generated by simulation by learning from the evolution of 16S rRNA genes can also be accurately reconstructed, and that the 16 million cells generated by simulation by learning from various cell engineering data ( The systematics of 16 million cells (equivalent to the number of cells in a 13.5-day embryo of a mouse) generated by simulation by learning various cell engineering data was also shown to be estimated with 99.5% accuracy. FRACTAL was also able to estimate reasonable genealogies not only for simulated data, but also for more than 3 million DNA sequences in which mutations were experimentally accumulated in vitro.
This method has the potential to significantly expand the horizon of evolutionary and developmental biology in the future. In evolutionary biology, phylogenetic analysis of metagenomic sequences obtained by comprehensive sequencing of DNA in the environment has recently revealed the existence of unknown microorganisms that are difficult to cultivate, and FRACTAL has the potential to comprehensively elucidate the phylogenetic relationships of new microbes and viruses FRACTAL has the potential to comprehensively elucidate the phylogenetic relationships of new microbes and viruses. FRACTAL is currently the only computational framework in the field of developmental biology that can analyze whole-body developmental processes of various animals at high resolution. It is expected to contribute to the elucidation of the whole-body developmental map of animals, which will lead to various new discoveries in biology.
Journals
-
Journal name Nature BiotechnologyTitle of paper Deep distributed computing to reconstruct extremely large lineage treesAuthor(s) Naoki Konno, Yusuke Kijima, Keito Watano, Soh Ishiguro, Keiichiro Ono, Mamoru Tanaka, Hideto Mori, Nanami Masuyama, Dexter Pratt, Trey Ideker, Wataru Iwasaki, and Nozomu Yachie*.DOI Number 10.1038/s41587-021-01111-2 Abstract URL https://www.nature.com/articles/s41587-021-01111-2
Terminology
Note 1 "Deep distributed computing" method FRACTAL
A computational method developed in this research for the estimation of large phylogenetic trees. It can incorporate existing phylogenetic tree estimation software as a plug-in, and the number of DNA sequences it can handle is greatly expanded by repeating distributed computing at multiple layers. ↑up
Note 2 Genome editing
A technique for precisely editing any sequence on a chromosome or converting it to a random sequence; the 2020 Nobel Prize in Chemistry was awarded to Dr. Janifer Doudna and Dr. Emmanuelle Charpentier for their development of this technique. ↑up