
DATE2021.05.20 #Press Releases
Genetic characterization of RNF213, a risk gene for moyamoya disease, and estimation of its diffusion pathway.
Disclaimer: machine translated by DeepL which may contain errors.
Yoshie Koganebuchi, Assistant Professor, Department of Biological Sciences
Hiroki Ota, Professor, Department of Biological Sciences
Key points of the presentation
- Population genetic analysis of RNF213, a risk gene for moyamoya disease, a disease with a high prevalence in East Asia, in Japanese patients showed that the diverse pathogenesis of this disease may be influenced not only by mutations in RNF213 but also by environmental factors, and that disease risk mutations appeared in continental East Asia in prehistoric times. The disease risk mutations emerged in continental East Asia during prehistoric times.
- Sequence analysis of the entire RNF213 gene in 24 patients with moyamoya disease revealed for the first time the population genetics of RNF213 in East Asia.
- The results of this study contribute to our understanding of the diversity of the pathogenesis of moyamoya disease.
Summary of Presentation
Moyamoya disease is a cerebrovascular disorder with a high prevalence in East Asian human populations. The disease is associated with a risk mutation R4810K in the RNF213 gene, which is observed only in East Asia. While there is a commonality in that approximately 90% of patients in Japan have this risk mutation, the symptoms of the disease are diverse.
A joint team (Kitasato University, University of the Ryukyus, Saga University, and The Institute of Statistical Mathematics) led by Professor Hiroki Ota and Assistant Professor Yoshie Koganebuchi of The University of Tokyo Graduate School of Science conducted a population genetics analysis based on the RNF213 gene sequences of Moyamoya disease patients.
The results suggest that the RNF213 gene with the risk mutation has a relatively recent origin in East Asia, and that the risk mutation originated in prehistoric continental East Asia and probably spread throughout the archipelago during the migration from the continent to the archipelago around the late Jomon Period (about 3,000 years ago). It has been suggested that this is the case.
Despite the high diversity of Moyamoya disease symptoms, the homogeneity of RNF213 sequences among patients suggests that the diversity of symptoms is due to the influence of environmental factors. The distribution of this risk variant is also associated with human migration history. Such population genetic analysis will contribute to our understanding of the pathogenesis of moyamoya disease.
Contents of Presentation
Background of the study and problems in previous studies
Moyamoya disease is one of the cerebrovascular disorders. In this disease, the terminus of the internal carotid artery, a large blood vessel in the brain, becomes narrow, which leads to blood shortage. To compensate for this, a new network of blood vessels is formed. Because these blood vessels look like fog, Japanese doctors have named the disease "moyamoya disease. The prevalence of patients with moyamoya disease has been reported to be higher among East Asians than in other parts of the world. In Japan, Moyamoya disease is recognized as a specific disease by the Ministry of Health, Labour and Welfare, and the number of patients is approximately 15,000. Moyamoya disease has a wide variety of forms, including cerebral ischemia, cerebral infarction, and cerebral hemorrhage, making it a life-threatening disease. About 10% of cases of Moyamoya disease occur in the same family. Therefore, genetic factors are thought to play a role in the development of Moyamoya disease, and several genomic regions related to the disease have been reported.
In 2011, two Japanese research teams independently reported the risk mutation gene RNF213 (Kamada et al. 2011; Liu et al. 2011) , which has been shown to be related to angiogenesis and to be a regulator of fat metabolism . The risk mutation for moyamoya disease is a Single Nucleotide Polymorphism (SNP) (Note 1) that changes the 4810th amino acid in the RNF213 protein from arginine to lysine, denoted R4810K. This risk mutation R4810K is known to exist almost exclusively in East Asia. While the frequency of non-moyamoya disease patients with this mutation is low, about 1% in each East Asian region, the frequency of the risk mutation in patients is highest in Japan at about 50%, followed by Korea at about 40% and China at about 10%. The risk of developing moyamoya disease in people with R4810K has also been reported to be particularly high in Japan.
In summary, while the symptoms of moyamoya disease are diverse, the risk mutation of a single gene has a strong influence on the disease. Therefore, it has been debated whether the variety of symptoms is due to environmental factors or whether another mutation different from R4810K may be affecting the symptoms. In this study, we characterized the RNF213 gene sequence in patients with and without moyamoya disease, and examined the timing of appearance of risk mutations and their diffusion process in East Asia.
Study Contents
To characterize the RNF213 gene sequence in patients with non-Moyamoya disease, haplotypes (Note 2) were estimated using genome sequences of a total of 414 individuals (108 African, 99 European, 103 Chinese, and 104 Japanese) from four populations in public databases. The number of haplotype types detected was higher in Africans than in non-Africans, which implies a higher diversity of the RNF213 gene in Africa. This result can be explained by the human history that the present humans (Homo sapiens) originated in Africa and spread from Africa to other parts of the world. It was also revealed that the Japanese genome in public databases contains a very small number of gene sequences containing risk mutations.
To characterize the RNF213gene sequences in patients with moyamoya disease who were admitted or attended Kitasato University Hospital, a phylogenetic tree was constructed using RNF213 gene sequences from 24 Japanese patients. As a comparison, gene sequences of 104 Japanese patients from a public database used in the previous haplotype analysis were also used. The results showed that most of the sequences with risk mutations were clustered together in one location in the phylogenetic tree (Figure 1).
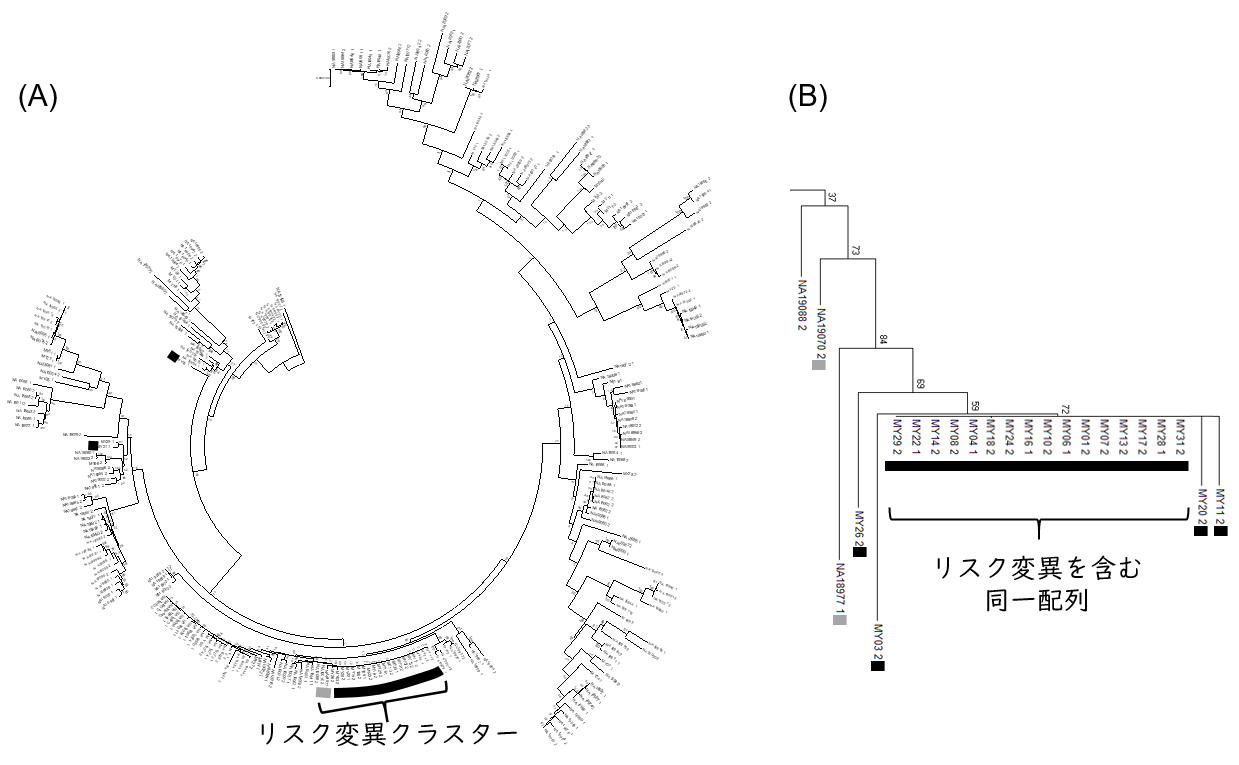
Figure 1: Phylogenetic tree of RNF213, a risk gene for moyamoya disease. The black marker contains RNF213 sequences containing risk variants derived from Moyamoya disease patients, and the gray marker contains RNF213 sequences containing risk variants derived from Japanese individuals in the public database who are non-Moyamoya disease patients. (A) Entire phylogenetic tree (B) Phylogenetic tree focusing on risk mutation clusters.
Focusing on this cluster of risk mutation sequences, interestingly, 80% of the 20 sequences derived from patients with Moyamoya disease were identical. In other words, the majority of the risk variant sequences were identical, even though the patients in this analysis were from different families. This suggests that the risk variant R4810K emerged in East Asia relatively recently in human history and has persisted to the present.
Based on the RNF213 gene sequence containing the mutation, the time of appearance of the risk mutation R4810K was estimated to be approximately 14,500-5,100 years ago (Figure 2). This age corresponds to the Jomon period in the Japanese archipelago. Anthropological and archaeological studies indicate that there was a large-scale human migration from East Asia to the Japanese archipelago about 3,000 years ago. In other words, it is suggested that risk mutations spread to the Japanese Islands as a result of this migration.
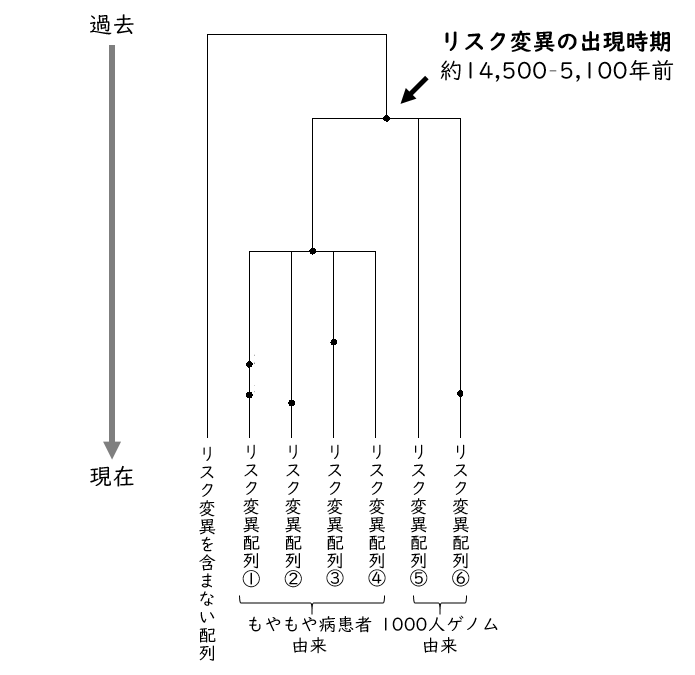
Figure 2: Genealogy of the RNF213 gene sequence inferred from simulations. Black dots indicate mutations that occurred in the DNA sequence.
Social Significance and Future Plans
The fact that the RNF213 gene sequences of the patients were almost identical suggests that the diverse pathogenesis of moyamoya disease is due to the influence of environmental factors rather than mutations in this gene. Although the influence of environmental factors has been pointed out previously, this study is the first to investigate risk genes at the sequence level and to support this point. In addition, infectious diseases, inflammatory diseases, autoimmune diseases, thyroid dysfunction, and abnormal lipid metabolism, which have been reported to be associated with moyamoya disease, are considered to be triggers, or environmental factors, for the development or exacerbation of moyamoya disease. The relationship between them requires further detailed investigation. The emergence of the moyamoya disease risk mutation is also thought to have occurred in a population that remained in the continental part of Japan after the Jomon people who migrated to the Japanese archipelago diverged from the ancestral population of all East Asians. The mutations may have come to the Japanese archipelago with the arrival of the Yayoi period and later, which began approximately 3,000 years ago. The results of this study will contribute to our understanding of the diversity of the pathogenesis of moyamoya disease by analyzing the risk mutations of the disease from a population genetic perspective.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Society for the Promotion of Science (JSPS), 24370099, 17H03738, 17H05132, 19H04526, 19H05350 (PI: Hiroki Ota), 16H06408 (PI: Hajime Ishida), 24300109 (PI: Shuhei Mano) The project was conducted with the support of
Journals
-
Journal Title Annals of Human Genetics Title of paper An analysis of the demographic history of the risk allele R4810K in RNF213 of moyamoya disease Author(s) Kae Koganebuchi, Kimitoshi Sato, Kiyotaka Fujii, Toshihiro Kumabe, Kuniaki Haneji, Takashi Toma, Hajime Ishida, Keiichiro Joh, Hidenobu Soejima, Shuhei Mano, Motoyuki Ogata, Shuhei Mano, Motoyuki Soejima Shuhei Mano, Motoyuki Ogawa, Hiroki Oota*, Shuhei Mano DOI Number 10.1111/ahg.12424 URL
Terminology
(Note 1) Single Nucleotide Polymorphism (SNP)
A single nucleotide polymorphism (SNP) is a difference between individuals in the human genome in which a single nucleotide is different at a specific location in the sequence with a frequency of 1% or more in a population. ↑(Note 1) SNP
(Note 2) Haplotype
Humans inherit one set of genome each from their father and mother. It refers to the sequence of genes derived from one of the parents. ↑ (Note 3)